1. Concatenate functions in caretSDM
[author name]
2026-07-24
Source:vignettes/articles/01_Compact.Rmd
01_Compact.RmdIn caretSDM we always use one central object through out the
framework, the input_sdm object. This allows us to
concatenate functions, which can be very useful when running SDMs for
the first couple of times. Here we provide a functioning way to easily
run your framework with only three objects.
# Open library
library(caretSDM)
#> Registered S3 methods overwritten by 'ggpp':
#> method from
#> heightDetails.titleGrob ggplot2
#> widthDetails.titleGrob ggplot2
set.seed(1)
# Build sdm_area object
sa <- sdm_area(rivs,
cell_size = 25000,
output_crs = 6933,
gdal = T,
lines_as_sdm_area = TRUE
) |>
add_predictors(bioc) |>
add_scenarios() |>
select_predictors(c("LENGTH_KM", "DIST_DN_KM", "bio1", "bio4", "bio12"))
#> ! Making grid over study area is an expensive task. Please, be patient!
#> ℹ Using GDAL to make the grid and resample the variables.
#> Linking to GEOS 3.12.1, GDAL 3.8.4, PROJ 9.4.0; sf_use_s2() is TRUE
#>
#> ! Making grid over the study area is an expensive task. Please, be patient!
#> ℹ Using GDAL to make the grid and resample the variables.
# Build occurrences_sdm object
oc <- occurrences_sdm(salm, occ_crs = 6933)
# Merge sdm_area and occurrences_sdm and perform pre-processing, processing and projecting.
i <- input_sdm(oc, sa) |>
data_clean() |>
multicollinearity_sdm("vifcor") |>
pseudoabsences(method = "bioclim", variables_selected = "vif") |>
train_sdm(
algo = c("naive_bayes", "kknn"),
ctrl = caret::trainControl(
method = "repeatedcv",
number = 4,
repeats = 1,
classProbs = TRUE,
returnResamp = "all",
summaryFunction = summary_sdm,
savePredictions = "all"
),
variables_selected = "vif"
) |>
predict_sdm(th = 0.7) |>
ensemble_sdm() |>
suppressWarnings()
#> Cell_ids identified, removing duplicated cell_id.
#> Testing country capitals
#> Removed 0 records.
#> Testing country centroids
#> Removed 0 records.
#> Testing duplicates
#> Removed 0 records.
#> Testing equal lat/lon
#> Removed 0 records.
#> Testing biodiversity institutions
#> Removed 0 records.
#> Testing coordinate validity
#> Removed 0 records.
#> Testing sea coordinates
#> Reading ne_110m_land.zip from naturalearth...Removed 0 records.
#> Loading required package: ggplot2
#> Loading required package: lattice
#> Ensemble function: average
#> current
i
#> caretSDM
#> ................................
#> Class : input_sdm
#>
#> =========== Overview ===========
#> Focal Taxon : Salminus brasiliensis
#> Spatial extent : -5269920.54541171, -3240235.69291754, -4700000, -2803133.9792114 (xmin,xmax,ymin,ymax)
#> Temporal extent : Current
#> Observation type : Presence-absence (pseudo-absence)
#> Predictor names : LENGTH_KM, DIST_DN_KM, bio1, bio4, bio12
#> Modelling algorithms : naive_bayes, kknn
#> Model complexity (tuneLength) : 1
#> Model averaging : average
#>
#> ============= Data =============
#> -- Biodiversity data --
#> Taxon names : Salminus brasiliensis
#> Sample size : 22
#> (Pseudo)Absence data method : bioclim
#> Number of PA sets : 10
#> PAs per set : 22
#> PA-to-presence ratio : 1
#> Data cleaning : NAs, Capitals, Centroids, Geographically Duplicated, Identical Lat/Long, Institutions, Invalid, Non-terrestrial, Duplicated Cell (grid)
#> -- Data partitioning --
#> Training/validation method : repeatedcv
#> Number of folds/repeats : 4
#> -- Predictor variables --
#> Number of predictors : 5
#> Predictor names : LENGTH_KM, DIST_DN_KM, bio1, bio4, bio12
#> Spatial extent : -5269920.54541171, -3240235.69291754, -4700000, -2803133.9792114 (xmin,xmax,ymin,ymax)
#> Spatial resolution : (25000, 25000)
#> Coordinate reference system : EPSG:6933 ( EPSG: 6933 )
#> -- Transfer data --
#> Number of scenarios : 1
#> Scenario names : current
#> Temporal extent : Current
#>
#> ============= Model ============
#> -- Multicollinearity --
#> Variable selection method : vif
#> Selected variables : LENGTH_KM, DIST_DN_KM, bio1
#> -- Model settings --
#> Predictors used : LENGTH_KM, DIST_DN_KM, bio1
#> Model hyperparameters :
#> species algorithm parameters
#> 1 Salminus brasiliensis naive_bayes_pa1 laplace=0, usekernel=FALSE, adjust=1
#> 2 Salminus brasiliensis kknn_pa1 kmax=7, distance=2, kernel=optimal
#> 3 Salminus brasiliensis naive_bayes_pa2 laplace=0, usekernel=TRUE, adjust=1
#> 4 Salminus brasiliensis kknn_pa2 kmax=9, distance=2, kernel=optimal
#> 5 Salminus brasiliensis naive_bayes_pa3 laplace=0, usekernel=TRUE, adjust=1
#> 6 Salminus brasiliensis kknn_pa3 kmax=5, distance=2, kernel=optimal
#> 7 Salminus brasiliensis naive_bayes_pa4 laplace=0, usekernel=TRUE, adjust=1
#> 8 Salminus brasiliensis kknn_pa4 kmax=9, distance=2, kernel=optimal
#> 9 Salminus brasiliensis naive_bayes_pa5 laplace=0, usekernel=TRUE, adjust=1
#> 10 Salminus brasiliensis kknn_pa5 kmax=9, distance=2, kernel=optimal
#> 11 Salminus brasiliensis naive_bayes_pa6 laplace=0, usekernel=TRUE, adjust=1
#> 12 Salminus brasiliensis kknn_pa6 kmax=9, distance=2, kernel=optimal
#> 13 Salminus brasiliensis naive_bayes_pa7 laplace=0, usekernel=FALSE, adjust=1
#> 14 Salminus brasiliensis kknn_pa7 kmax=9, distance=2, kernel=optimal
#> 15 Salminus brasiliensis naive_bayes_pa8 laplace=0, usekernel=TRUE, adjust=1
#> 16 Salminus brasiliensis kknn_pa8 kmax=5, distance=2, kernel=optimal
#> 17 Salminus brasiliensis naive_bayes_pa9 laplace=0, usekernel=FALSE, adjust=1
#> 18 Salminus brasiliensis kknn_pa9 kmax=9, distance=2, kernel=optimal
#> 19 Salminus brasiliensis naive_bayes_pa10 laplace=0, usekernel=TRUE, adjust=1
#> 20 Salminus brasiliensis kknn_pa10 kmax=9, distance=2, kernel=optimal
#> -- Threshold selection --
#> Threshold method : threshold
#> Threshold criteria : 0.7
#>
#> ========== Assessment ==========
#> -- Performance statistics --
#> Cross-validation metrics :
#> $`Salminus brasiliensis`
#> algo ROC TSS Sensitivity Specificity
#> 1 kknn 0.7600972 0.3641667 0.736625 0.63165
#> 2 naive_bayes 0.7784722 0.4150000 0.760850 0.67335
#>
#>
#> ========== Prediction ==========
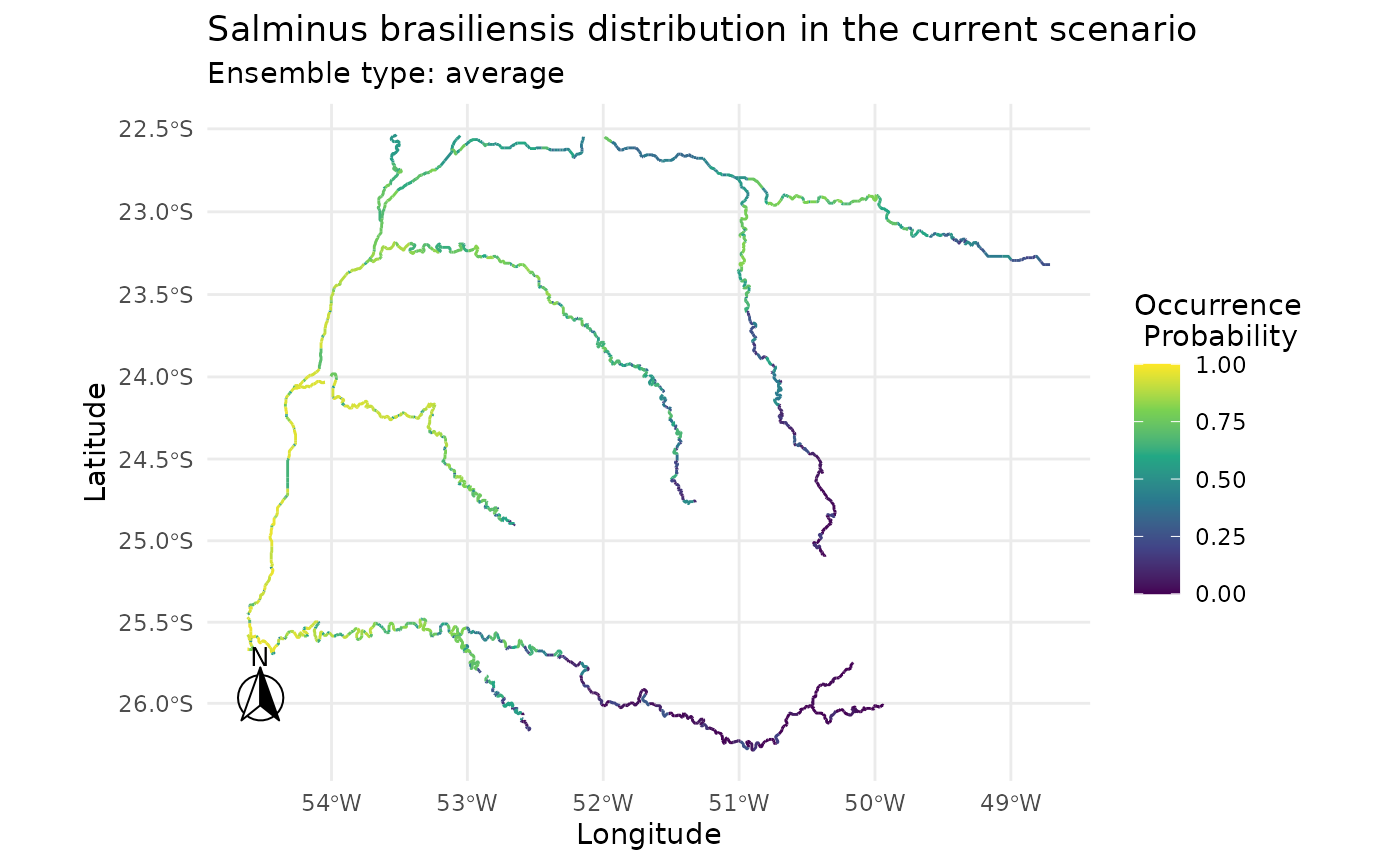
#> Prediction layers : current
#> Prediction unit : Occurrence Probability
#> Ensemble method : average
#> Ensemble names :
plot(i)